

[GROMACS]模拟数据分析前轨迹文件生成-轨迹预处理
前言
在gromacs动力学中,原始轨迹文件(.xtc/.trr)因周期性边界条件(PBC)和整体平动/转动的存在,无法直接用于定量分析或可视化。本文介绍三类核心预处理方法,分别针对连续轨迹、结构叠合和可视化需求,并明确每类输出文件的适用分析场景与注意事项。为了得到一个用于可视化和适配于各种数据分析使用的轨迹,我们需要对轨迹进行三种预处理,用于不同的数据分析和可视化。
| 预处理类型 | 输出文件示例 | 核心目标 | 典型应用 |
|---|---|---|---|
| PBC跳跃消除 | md_nojump.xtc |
还原真实连续运动,保留绝对坐标 | MSD、RDF、能量rerun |
| 整体运动叠合 | md_fit.xtc |
固定蛋白质,消除平动/转动 | RMSD、RMSF、氢键、DCCM |
| 可视化优化 | md_center.xtc |
画面整洁,分子完整居中 | 轨迹动画展示 |
注意:尽量保留原始轨迹!每次处理生成新文件,不要覆盖,保证预处理逻辑更清晰
一、PBC 跳跃消除:获得连续轨迹(md_nojump.xtc)
周期性边界条件 (PBC):在分子动力学模拟中,为了模拟一个无限大的体系,我们使用一个有限的盒子,并让这个盒子在三个维度上无限重复。当一个分子(比如你的小分子配体)从盒子的一侧穿出去时,它会立刻从盒子的另一侧穿进来。这在计算上是连续的,但在轨迹文件中记录的坐标会发生一个巨大的跳跃(从盒子的一端到另一端)。
1.1命令与参数详解
gmx trjconv -s md.tpr -f md.xtc -n index.ndx -o md_nojump.xtc -pbc nojump| 参数 | 含义 | 详细说明 |
|---|---|---|
-s md.tpr |
结构拓扑/输入文件 | 包含盒子信息、原子质量、键连接等。-s 通常指.tpr文件,提供参考坐标和盒子。 |
-f md.xtc |
输入轨迹 | 原始轨迹,可以是.xtc(压缩)或.trr(全精度)。 |
-n index.ndx |
索引文件 | 可选推荐。定义原子组(如 Protein、Ligand、Backbone),用于后续选择参考组。若没有,gmx trjconv会交互式提示你从预定义组中选择。 |
-o md_nojump.xtc |
输出轨迹 | 处理后的连续轨迹。 |
-pbc nojump |
核心参数 | “No Jump”:检测并消除分子因PBC产生的跳跃。算法:对于每个分子,追踪其质心轨迹,当质心跨过盒子边界时,将该分子的所有原子坐标平移回连续位置。 |
-pbc nojump 的底层原理:假设盒子长度为 10 nm,一个分子在第 10 帧坐标 x = 9.9 nm,第 11 帧从右侧穿出,实际物理位置应为 x = 10.1 nm,但由于PBC,轨迹记录为 x = 0.1 nm。-pbc nojump 会检测到跳跃 > 半个盒长(默认阈值 0.45×盒长),自动给该分子所有原子加上 ±L 使其连续。最终第 11 帧被修正为 10.1 nm。
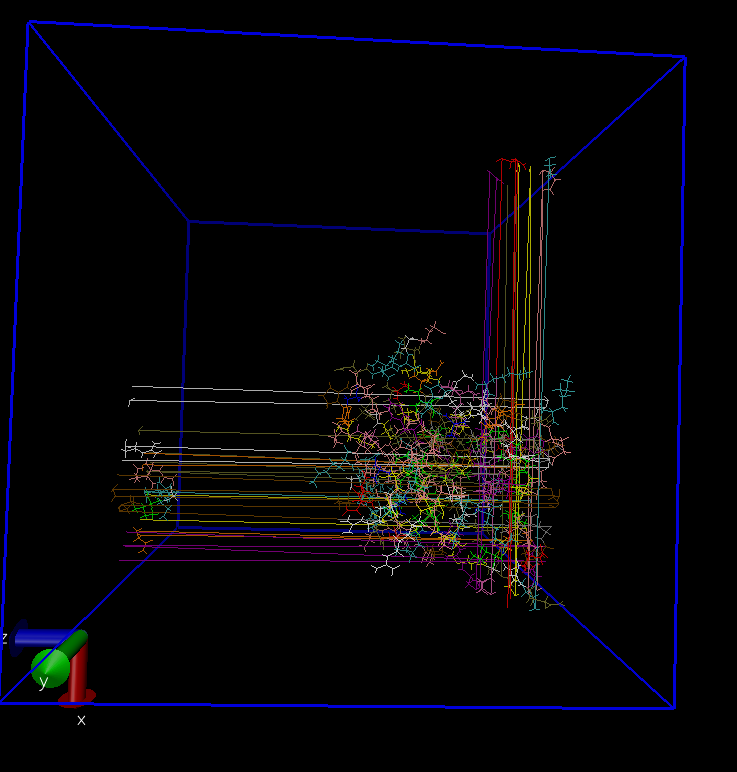
如图上图为消除跳跃之前的,会发现超出PBC盒子以外的部分发生跳跃从盒子对侧出现,而当我们消除之后如下图,你会发现,即便超出盒子区域,仍然不会发生跳跃
重要:
-pbc nojump依赖分子完整性。若分子本身在轨迹中被“切开”(例如配体的一部分在盒子一边,另一部分在对面),则需要先用-pbc whole将分子拼接完整。
1.2 前置步骤:确保分子完整(-pbc whole)
什么时候必须用 -pbc whole?
当模拟中分子跨过了盒子边界,且轨迹记录时没有保持分子完整性(默认GROMACS输出的轨迹中,原子坐标按照其在盒子中的真实位置记录,一个分子可能被“劈”成两半分别位于盒子两端)。这种情况常见于:
- 长链分子(如聚合物、某些配体)
- 膜蛋白中的脂质分子
- 使用
-pbc nojump前没有做任何处理
如图为被劈成两半的氨基酸分子。
解决方法:先执行一次 -pbc whole:
gmx trjconv -s md.tpr -f md.xtc -n index.ndx -o md_whole.xtc -pbc whole-pbc whole:将每个分子的所有原子拼回同一盒子内,通过搜索每个原子的周期性映像,使得分子内原子间距离最小化(即分子不断裂)。- 之后再对
md_whole.xtc执行-pbc nojump。
一条命令同时处理 whole + nojump(管道,避免中间文件):
gmx trjconv -s md.tpr -f md.xtc -n index.ndx -o md_nojump.xtc -pbc whole -pbc nojump注意:多个 -pbc 参数按顺序执行,先 whole 后 nojump。
1.4 PBC跳跃消除轨迹-适用分析
| 分析 | 是否可用 | 备注与必要条件 |
|---|---|---|
| 均方位移(MSD) | 必须 | 需要绝对坐标的连续性。使用 gmx msd 时,必须输入 -pbc nojump 处理后的轨迹。 |
| 径向分布函数(RDF) | 必须 | 执行 gmx rdf 时,必须加上 -pbc no,因为轨迹已经消除了跳跃,无需再搜索周期性映像。 |
| 能量 rerun(如相互作用能) | 可用 | 前提:轨迹中分子完整(已用 -pbc whole)。若跳过 whole,能量计算可能错误(原子位置错误)。 |
| 回旋半径(Rg) | 完美 | 无需叠合,连续轨迹即可。 |
| 氢键寿命 | 可用 | 依赖连续时间序列,但不依赖绝对位置。 |
| 自相关函数(ACF) | 可用 | 同上。 |
| 扩散系数(Einstein relation) | 必须 | MSD 的子项。 |
| PCA(主成分分析) | 不可 | PCA 需要先去除整体平动/转动,否则前几个主成分为整体运动。 |
| RMSD / RMSF | 不可 | 除非后续用 gmx rms 的 -fit 选项叠合,但最好单独做叠合轨迹。 |
| DCCM / 动力学相关性 | 不可 | 需要去除整体运动。 |
| 可视化(VMD/PyMOL) | 勉强 | 分子可能不在盒子中央,且可能仍有残留跳跃(如果 -pbc nojump 未能完全处理某些情况)。 |
二、整体平动与转动消除:结构叠合轨迹(md_fit.xtc)
此处是为了消除动力学模拟中发生的平动和转动,消除后体系数据分析更稳定
2.1 命令与参数详解
gmx trjconv -s md.tpr -f md_nojump.xtc -n index.ndx -o md_fit.xtc -fit rot+trans -center参数拆解
| 参数 | 含义 | 详细说明 |
|---|---|---|
-fit rot+trans |
叠合方式 | rot 旋转 + trans 平动。将每一帧的指定参考组(通常是蛋白质骨架)通过最小二乘拟合,叠合到第一帧(默认参考帧)。 |
-center |
居中 | 将指定组(通常是蛋白质)平移到盒子中心。通常与 -fit 配合使用。 |
注意:-fit 和 -center 都需要交互选择组:
- Least-squares fit group:选择叠合参考组(如
Backbone或C-alpha)。这个组的原子坐标将被用来计算旋转和平动矩阵。 - Output group:输出哪个组?通常选择
System(所有原子),这样整个体系(包括配体、水、离子)都随蛋白质一起旋转平移。 - Centering group(如果用了
-center):选择居中的组,一般也是Protein。
先做 -pbc nojump 再做叠合原因
如果直接用原始轨迹做叠合,由于 PBC 跳跃,第 10 帧蛋白质在盒子中央,第 11 帧蛋白质可能“跳”到盒子角落,叠合算法会错误地将整体平移很大距离,导致配体位置错乱。必须先消除跳跃,使轨迹在绝对坐标下连续,再做叠合。
2.2 叠合的底层原理:最小二乘拟合
每一帧的原子坐标集合 {r_i},参考帧 {r_i^ref}。寻找旋转矩阵 R 和平移向量 t,使得下式最小:
∑ w_i |R·r_i + t - r_i^ref|^2 其中 w_i 通常是原子质量(默认)或等权重。GROMACS 使用 Kabsch 算法。只对 fit group 计算 R,t,然后应用到整个 output group。
2.4 适用分析
| 分析 | 是否可用 | 备注 |
|---|---|---|
| RMSD(均方根偏差) | 完美 | 直接 gmx rms -s md.tpr -f md_fit.xtc |
| RMSF(涨落) | 完美 | 先做 RMSD 拟合轨迹,或用 gmx rmsf -res |
氢键数量(gmx hbond) |
完美 | 依赖相对距离和角度,不受整体运动影响 |
盐桥(gmx saltbr) |
完美 | 同上 |
溶剂可及表面积(gmx sasa) |
完美 | 依赖构象,与整体运动无关 |
| 动态交叉关联矩阵(DCCM) | 必须 | DCCM 要求去除整体运动,否则虚假相关 |
| 自由能形貌图(FEL) | 完美 | 基于 PCA 或 RMSD 等内部坐标 |
| 二级结构(DSSP) | 完美 | gmx do_dssp 基于局部几何 |
平均结构(gmx covar + gmx anaeig) |
完美 | 必须先叠合轨迹 |
| 主成分分析(PCA) | 必须 | 必须去除整体运动 |
| MM/PBSA 结合自由能 | 谨慎 | 若使用 gmx mmgbsa,需要连续轨迹且无整体运动,但需注意:MM/PBSA 通常使用无溶剂轨迹(仅蛋白+配体),且需要确保坐标与拓扑一致。建议对 md_fit.xtc 提取蛋白+配体子集。 |
| MSD | 不可 | 绝对坐标已被改变 |
| RDF | 不可 | 相对位置虽保留,但盒子信息可能被破坏(-center 平移后盒子不变?GROMACS 中盒子矢量不变,仅原子坐标平移,RDF 理论上仍正确?实际上 -center 不改变相对距离,但很多用户担心,所以建议 RDF 用 md_nojump.xtc)。 |
| 能量 rerun | 危险 | 坐标可能超出原始盒子边界(虽然盒子矢量没变,但平移后部分原子可能跑到盒子外),PME 计算可能出错。 |
2.6 错误问题
- 错误1:叠合时选错了输出组。如果输出组只选了蛋白质,配体就没有被旋转平移,配体会相对于蛋白质“飞走”。必须选
System。 - 错误2:参考组包含了配体或柔性 loop。配体运动会导致叠合扭曲,RMSD 虚高。
- 错误3:没有先做
-pbc nojump直接叠合。蛋白质的跳跃会导致叠合失败,轨迹中出现异常跳跃。 - 错误4:使用
-fit rot+trans但忘记-center。虽然没有原则错误,但蛋白质可能不在盒子中央,可视化不美观。不过对 RMSD 等无影响。
三、可视化优化:最优轨迹(md_center.xtc)
3.1 命令与参数详解
gmx trjconv -s md.tpr -f md.xtc -n index.ndx -o md_center.xtc -pbc mol -center -ur compact参数详解
| 参数 | 含义 | 详细说明 |
|---|---|---|
-pbc mol |
分子完整化 | 将每个分子的所有原子通过周期性平移放到同一盒子内(类似 -pbc whole,但不改变质心位置,只确保分子内原子不断裂)。 |
-center |
居中 | 将指定组(通常是蛋白质)平移到盒子中心。 |
-ur compact |
使用紧凑盒子表示 | 重新定义盒子矢量,使得盒子刚好包裹所有原子(最小化空白区域)。这改变了盒子的形状和大小,因此会改变原子间的相对距离! |
注意:-ur compact 会修改盒子尺寸,从而改变分子间的距离!例如,如果盒子原来是立方体 10×10×10 nm,但所有原子聚集在中央一个 3×3×3 区域,-ur compact 会将盒子缩小到刚好包裹所有原子(可能变成 3.2×3.2×3.2 nm)。这将导致:
- 周期性的距离计算错误(因为盒子变小,周期性映像更近)
- 对于多分子体系,分子间的相对距离(跨盒子)被破坏
因此,此轨迹最好不要用于任何定量分析!
3.2 可视化需要的操作
- 原始轨迹中,蛋白质可能不在中心,且水分子散落各处,看起来杂乱。
-pbc mol消除了断裂的分子(如某水分子一半在左一半在右)。-center让蛋白质居中,便于观察。-ur compact移除大片空白区域,视频文件更小,渲染更快。
3.3 适用场景(仅限视觉用途)
| 用途 | 说明 |
|---|---|
| 制作模拟动画(VMD/PyMOL) | 画面整洁,聚焦蛋白质-配体。 |
| 提取首尾帧做构象对比图 | 例如论文中的 Figure 1:模拟前后叠合图。 |
| 粗略观察配体结合模式变化 | 仅定性判断,不可计算距离。 |
3.4 验证与注意事项
验证:在 VMD 中加载,打开“Periodic”显示(如果 VMD 能显示盒子),你会看到盒子缩小了。
检查:运行 gmx check -f md_center.xtc,查看盒子尺寸,应该远小于原始盒子(如果体系浓缩)。
重要:如果你想在可视化中保留正确的相对距离(例如配体与蛋白质的距离看起来不变形),不要使用 -ur compact,只使用 -pbc mol -center:
gmx trjconv -s md.tpr -f md.xtc -n index.ndx -o md_visual_noCompact.xtc -pbc mol -center这样盒子保持不变,仅居中,距离信息保留。但注意,即使如此,这个轨迹仍不适合定量分析,因为没有去除整体运动(蛋白质可能在转动)。
参考文献
- GROMACS 官方手册:
gmx trjconv章节,https://manual.gromacs.org/current/onlinehelp/gmx-trjconv.html - “Handling periodic boundary conditions” – GROMACS Tutorials by Justin Lemkul
- “Best practices for analysis of MD trajectories” – BioExcel CoE
本文遵循 CC BY-NC 4.0 许可,欢迎转载但请注明出处。

