

[MMPBSA]深入解读 gmx_MMPBSA 计算结果:从焓、熵到残基分解的结构化解析
在使用 gmx_MMPBSA 完成结合自由能计算后,gmx_MMPBSA_ana 会输出一个树形结构的结果汇总。该结构严格按照热力学函数的构成进行组织,涵盖了焓变(ΔH)、熵变贡献(-TΔS)、结合自由能(ΔG)以及基于残基的能量分解。本文我们将以一个同时包含 GB 和 PB 计算、并启用相互作用熵与残基分解的典型输入文件为例,逐一解释结果树中每一项的物理意义及其背后的理论依据。
我们将用如下的一个基础分享配置输入文件来讲解。
# =======================================================
# 综合 mmpbsa_gen.in 文件
# 包含:GB、PB、能量分解、丙氨酸扫描、相互作用熵
# 力场:蛋白质 ff14SB,小分子 GAFF
# gmx_MMPBSA v1.6.4
# =======================================================
# 通用部分
&general
sys_name = "Protein_Ligand" # 系统名称(用于分析)
startframe = 9991 # 起始帧
endframe = 10001 # 结束帧
interval = 1 # 取帧间隔(10帧取1,降低计算量)
forcefields = "leaprc.protein.ff14SB,leaprc.gaff" # 力场:蛋白质ff14SB + 小分子GAFF
PBRadii = 3 # PB半径集(3 = mbondi2,常用)
temperature = 298.15 # 温度(K)
verbose = 2 # 输出详细程度
keep_files = 1 # 保留临时文件
netcdf = 1 # 使用NetCDF加速
interaction_entropy= 1 # 开启相互作用熵
ie_segment = 25 # 用最后25%的帧计算熵
/
# GB 计算
&gb
igb = 5 # GB模型(5 = GB‑OBC2)
saltcon = 0.150 # 盐浓度(M)
intdiel = 1.0 # 溶质介电常数
extdiel = 78.5 # 溶剂介电常数
surften = 0.0072 # 表面张力(kcal/mol/Ų)
probe = 1.4 # 探针半径(Å)
/
# PB 计算
&pb
istrng = 0.150 # 盐浓度(M)
fillratio = 4.0 # 网格填充比例
radiopt = 1 # 使用Tan‑Luo半径
inp = 2 # 非极性项分解(2 = 空腔+色散)
cavity_surften = 0.0378 # 空腔表面张力
cavity_offset = -0.5692 # 空腔偏移
use_sav = 1 # 用体积计算空腔项
decompopt = 2 # 非极性分解方案
use_rmin = 1 # 用rmin半径
/
# 能量分解
&decomp
idecomp = 2 # 2 = 残基分解(1‑4 EEL加到EEL,1‑4 VDW加到VDW)
dec_verbose = 1 # 1 = 输出总贡献
print_res = "within 5" # 输出距离结合界面5 Å以内的残基
csv_format = 1 # 输出CSV文件,方便画图
/
# 丙氨酸扫描(可以加入,在酶活改造领域至关重要,目前我们先不了解,后面我们单独详细介绍)
#&alanine_scanning
# mutant = "ALA" # 突变成丙氨酸
# mutant_res = "A:45" # 要突变的残基(链A,45号),需根据实际修改!!!!!!!!
# mutant_only = 0 # 0 = 同时计算突变型和野生型
# cas_intdiel = 1 # 根据残基极性自动调整介电常数
# intdiel_nonpolar = 1 # 非极性残基介电常数
# intdiel_polar = 3 # 极性残基介电常数
# intdiel_positive = 5 # 正电残基介电常数
# intdiel_negative = 5 # 负电残基介电常数
#/
1. 基本原理与计算框架
MMPBSA(Molecular Mechanics Poisson–Boltzmann Surface Area)和 MMGBSA(Generalized Born Surface Area)是基于连续介质模型的自由能计算方法。其核心假设是结合自由能可以分解为气相分子力学能量、溶剂化自由能以及熵贡献的统计平均值:
\Delta G_{\text{bind}} = \langle G_{\text{complex}} \rangle - \langle G_{\text{receptor}} \rangle - \langle G_{\text{ligand}} \rangle
其中各组分的自由能由下式给出:
G = E_{\text{MM}} + G_{\text{solv}} - TS
E_{\text{MM}}为分子力学能量,包括成键项(键伸缩、角弯曲、二面角)和非键项(范德华、静电)。G_{\text{solv}}为溶剂化自由能,进一步分为极性部分(G_{\text{pol}})和非极性部分(G_{\text{nonpol}})。S为溶质构型熵,通常通过正则模分析或准谐波分析获得,在 gmx_MMPBSA 中也可使用相互作用熵(Interaction Entropy, IE)近似。
由于成键项在单点能计算中几乎不变,其对结合能的净贡献(Δ)接近于零,因此最终关注的能量项主要来自非键相互作用与溶剂化效应的差值。
1.1溶剂化自由能的极性部分:PB 模型与 GB 模型
在 MMPBSA/MMGBSA 的框架中,溶剂化自由能 G_{\text{solv}} 被拆分为极性 G_{\text{pol}} 与非极性 G_{\text{nonpol}} 两部分。其中,非极性项通常正比于溶剂可及表面积(SASA),用于描述溶质-溶剂间的疏水与范德华相互作用;而极性项则源于溶质分子与极性溶剂(水)之间的静电相互作用——这一项的计算正是 PB 与 GB 模型的核心区别所在。
Poisson–Boltzmann(PB)模型是一种基于连续介质电静力学理论的严格方法。它将溶剂视为具有均匀介电常数(通常设为 80 左右)的连续介质,而溶质内部则采用较低的介电常数(常取 2–4)。在给定溶质原子电荷分布的情况下,静电势 \phi(\mathbf{r}) 满足 Poisson 方程:
\nabla \cdot [\varepsilon(\mathbf{r}) \nabla \phi(\mathbf{r})] = -4\pi \rho(\mathbf{r})
若溶液中存在可移动的离子(如生理盐环境),则需引入 Boltzmann 分布来描述离子云的空间分布,此时方程变为非线性 Poisson–Boltzmann 方程:
\nabla \cdot [\varepsilon(\mathbf{r}) \nabla \phi(\mathbf{r})] - \bar{\kappa}^2 \sinh\left(\frac{e\phi}{k_B T}\right) = -4\pi \rho(\mathbf{r})
其中 \bar{\kappa} 为修正的德拜屏蔽参数。通过数值求解(有限差分或有限元法),可获得溶质周围完整的电势分布,进而计算出将溶质从真空“充入”连续溶剂所需的极性溶剂化自由能:
G_{\text{pol}}^{\text{PB}} = \frac{1}{2} \sum_i q_i \left[ \phi(\mathbf{r}_i) - \phi_{\text{vac}}(\mathbf{r}_i) \right]
PB 模型的优势在于严格处理分子形状、电荷分布以及离子屏蔽效应,尤其适用于高度带电或形状不规则的大分子体系。然而,其数值求解的计算代价较高,限制了在高通量或长时程采样中的应用。
Generalized Born(GB)模型则是一种近似但高效的替代方案。它基于 Born 离子溶剂化自由能的经典公式推广而来:对于任意形状的分子,第 i 个原子的极性溶剂化自由能可写为:
G_{\text{pol, i}}^{\text{GB}} = -\frac{1}{2}\left(1 - \frac{1}{\varepsilon_w}\right) \frac{q_i^2}{a_i}
其中 a_i 为 Born 半径,代表该原子到分子表面的有效电介质距离。当体系中存在多个原子时,必须考虑相互屏蔽效应,因此总极性项通过成对求和近似得到:
G_{\text{pol}}^{\text{GB}} \approx -\frac{1}{2}\left(1 - \frac{1}{\varepsilon_w}\right) \sum_{i,j} \frac{q_i q_j}{f_{ij}^{\text{GB}}(r_{ij}, a_i, a_j)}
f_{ij}^{\text{GB}} 是一个经验函数(如 Still、HCT、OBC 等不同形式),当 r_{ij} 较大时退化为库仑相互作用(被溶剂介电常数屏蔽),当 r_{ij} 很小时则趋近于 Born 极限。GB 模型不需要求解三维偏微分方程,而是通过解析近似快速估算每个原子的有效 Born 半径,再累加得到总极性溶剂化能。常见的 GB 变体(如 GB-OBC、GB-neck2)通过经验参数修正,能在许多体系中与 PB 结果达到良好的一致性(偏差通常小于 1–2 kcal/mol),而计算速度却快 1–2 个数量级。
PB 与 GB 的权衡:在实际的 MMPBSA/MMGBSA 计算中,选择 PB 还是 GB 取决于精度与效率的平衡。PB 严格物理,适合精细研究(如电荷突变、离子强度影响);GB 计算廉价,适合大规模采样或作为力场再加权的一部分。值得注意的是,无论是 PB 还是 GB,其计算的都是静电溶剂化自由能,而最终的结合自由能还需要与气相分子力学非键项 \Delta E_{\text{vdw}} + \Delta E_{\text{elec}} 协同考虑——因为溶剂化能中的极性部分天然地补偿了气相静电相互作用的高估。这种“气相静电 + 极性溶剂化”的配对构成了连续介质模型中经典的“库仑 + Born”机制,也是理解结合自由能热力学来源的关键视角。
2. Enthalpy (ΔH) 部分:气相与溶剂化能量的分解
结果树中 Enthalpy 标题下展开的 Normal 子项,代表标准未突变的焓计算。此处同时列出 GB 和 PB 两套结果,二者仅在溶剂化模型上存在差异。
2.1 Complex、Receptor、Ligand 下的能量项
对于每一帧轨迹,程序分别计算复合物、受体和配体的单点能量,并在每个组分下列出相同的能量项。以 GB 下的 Complex 为例:
- BOND:键伸缩能。从力场参数计算,因坐标冻结成键项基本恒定。
- ANGLE:角弯曲能。同样几乎不变。
- DIHED:二面角扭转能。贡献极小且几乎在差分中抵消。
- VDWAALS:范德华相互作用能,由 Lennard-Jones 势描述。反映原子间的色散与交换排斥,通常为负值(有利),是结合焓的主要来源之一。
- EEL:真空静电相互作用能(库仑势)。在介电常数为 1 的真空中计算,数值往往很大。
- 1-4 VDW 与 1-4 EEL:1-4 非键相互作用,即被共价键间隔三个键的原子对之间的范德华和静电作用。力场中这些项的缩放因子通常小于 1,其贡献单独列出,并在 Delta 中抵消或调整。
- EGB:广义 Born 模型计算的极性溶剂化自由能。该能量反映了溶质电荷分布在溶剂中引起的反应场能量,是补偿真空静电作用的关键项。输入文件中
igb=5表示采用 GB-OBC2 模型,intdiel=1.0和extdiel=78.5分别定义了溶质内部与溶剂的介电常数。 - ESURF:非极性溶剂化自由能。通过溶剂可及表面积(SASA)乘以表面张力常数计算,此处
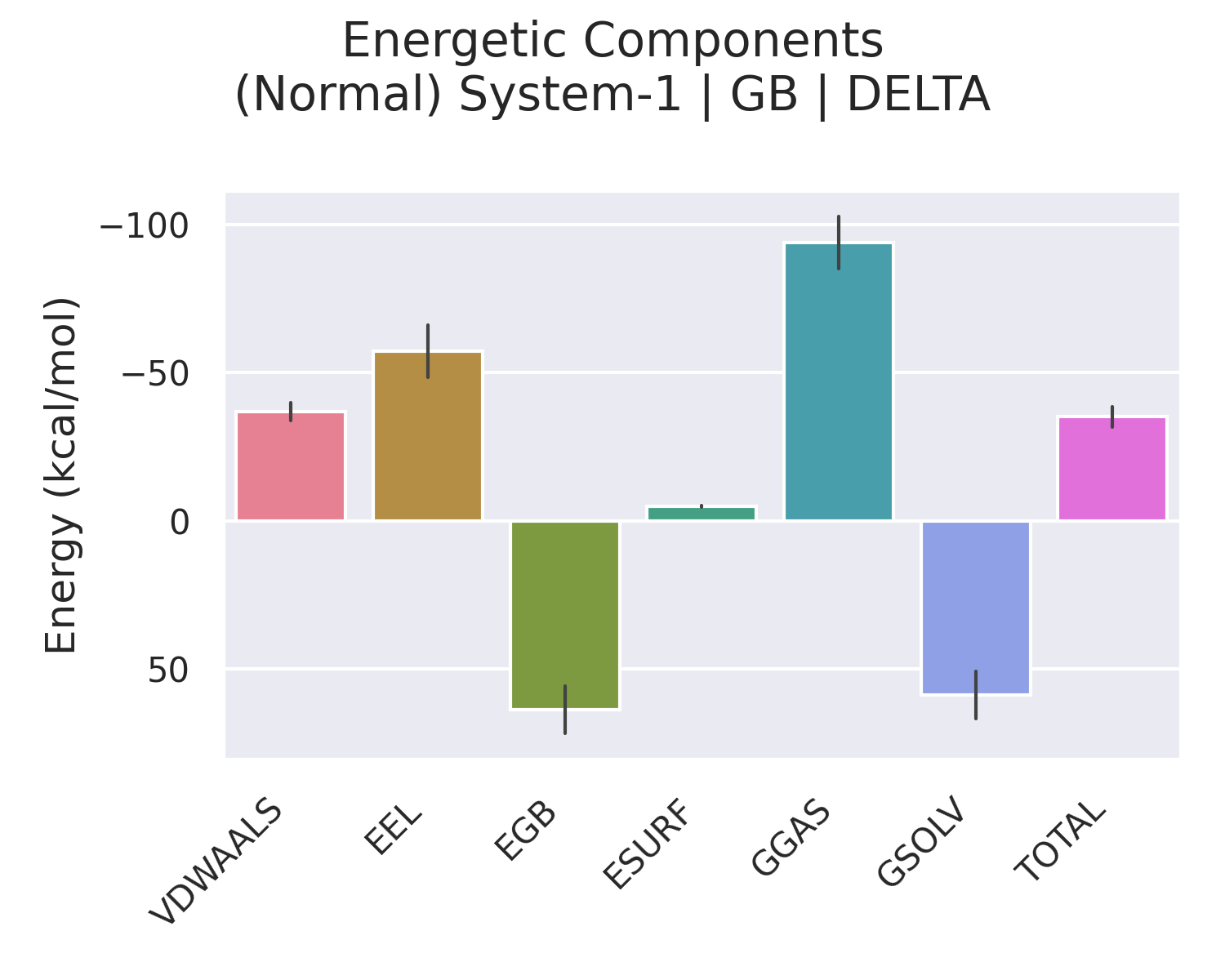
surften=0.0072 kcal/mol/Ų。非极性溶剂化项主要模拟空穴形成能与溶质-溶剂色散作用,通常为负,对疏水结合有重要贡献。 - GGAS:气相总能量,定义为
VDWAALS + EEL(有时也包括 1-4 项,取决于分解设置),表示除去溶剂效应的分子内与分子间非键能。 - GSOLV:溶剂化总自由能,即
EGB + ESURF(PB 下为EPB + ENPOLAR)。 - TOTAL:该组分在溶剂中的总势能,即
GGAS + GSOLV(或包含成键项后的总和)。此值并非结合焓,需进一步求差。
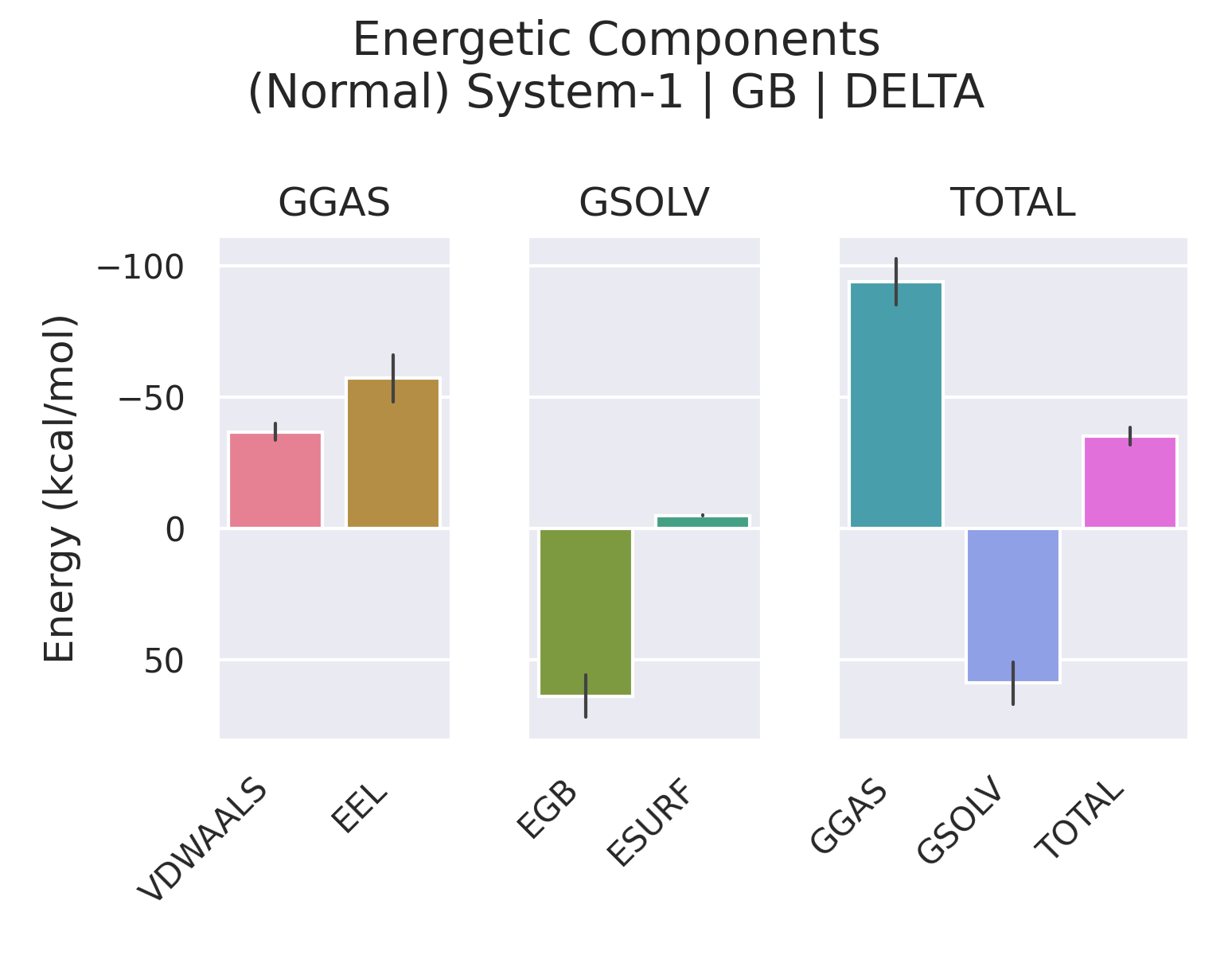
对于 PB 模型,上述 EGB 被替换为 EPB(Poisson–Boltzmann 静电能),ESURF 可能被更精细的非极性项(如空穴项与色散项)取代。我们上面的输入文件中 inp=2 指定了非极性项包含空穴与色散贡献,同时 cavity_surften 和 cavity_offset 控制空穴项计算。PB 通常被认为比 GB 更精确,但计算量更大。
2.2 Delta:结合引起的各项能量变化
Delta 分组下的每一项数值,均由 Complex - (Receptor + Ligand) 计算得到。例如:
- ΔVDWAALS:复合物形成时范德华相互作用的变化。负值表示结合后受体与配体间的范德华接触增强,是结合焓的重要驱动力。
- ΔEEL:真空静电相互作用变化。通常为负(有利),但会被 ΔEGB 部分抵消。
- ΔEGB:极性溶剂化的变化。受体与配体结合时部分极性基团去溶剂化,溶剂化能升高(正 ΔEGB),从而抵消部分真空静电收益。ΔEEL + ΔEGB 的和反映净静电贡献。
- ΔESURF:非极性溶剂化的变化。结合时溶剂可及表面积减少,ΔESURF 为负,提供疏水自由能贡献。
- ΔTOTAL:结合焓 ΔH 的估计值。在标准的 MMPBSA 流程中,它就是结合自由能计算中 ΔH 的部分。
因此,Enthalpy 部分的 Delta → TOTAL 正是所需要的 ΔH(对于 GB 或 PB)。
3. Entropy (-TΔS) 部分:相互作用熵的解读
我们上面的输入文件中设置了 interaction_entropy=1,因此结果中熵变部分显示为 IE,而非 NMODE。IE 方法从 MD 轨迹中直接估算熵变,其原理基于配体-受体相互作用能涨落与结合熵之间的关系:
-T\Delta S \approx kT \ln\langle e^{\beta \Delta E^{\text{int}}(t)} \rangle
其中 \Delta E^{\text{int}} 是配体与受体之间的相互作用能(气相下),尖括号表示系综平均。ie_segment=25 表示采用轨迹最后 25% 的帧进行涨落分析。
IE 方法得到的 -TΔS 会与选用的焓计算模型配对,因此你看到 Entropy → IE 下同时有 GB 和 PB 两个数值,这并不代表 IE 本身依赖溶剂模型,而是表明它将被分别加和到 GB 或 PB 的 ΔH 上以形成最终的结合自由能。通常 IE 计算的熵贡献对结合不利(-TΔS 为正),因为结合过程限制了配体的平动、转动与构象自由度。
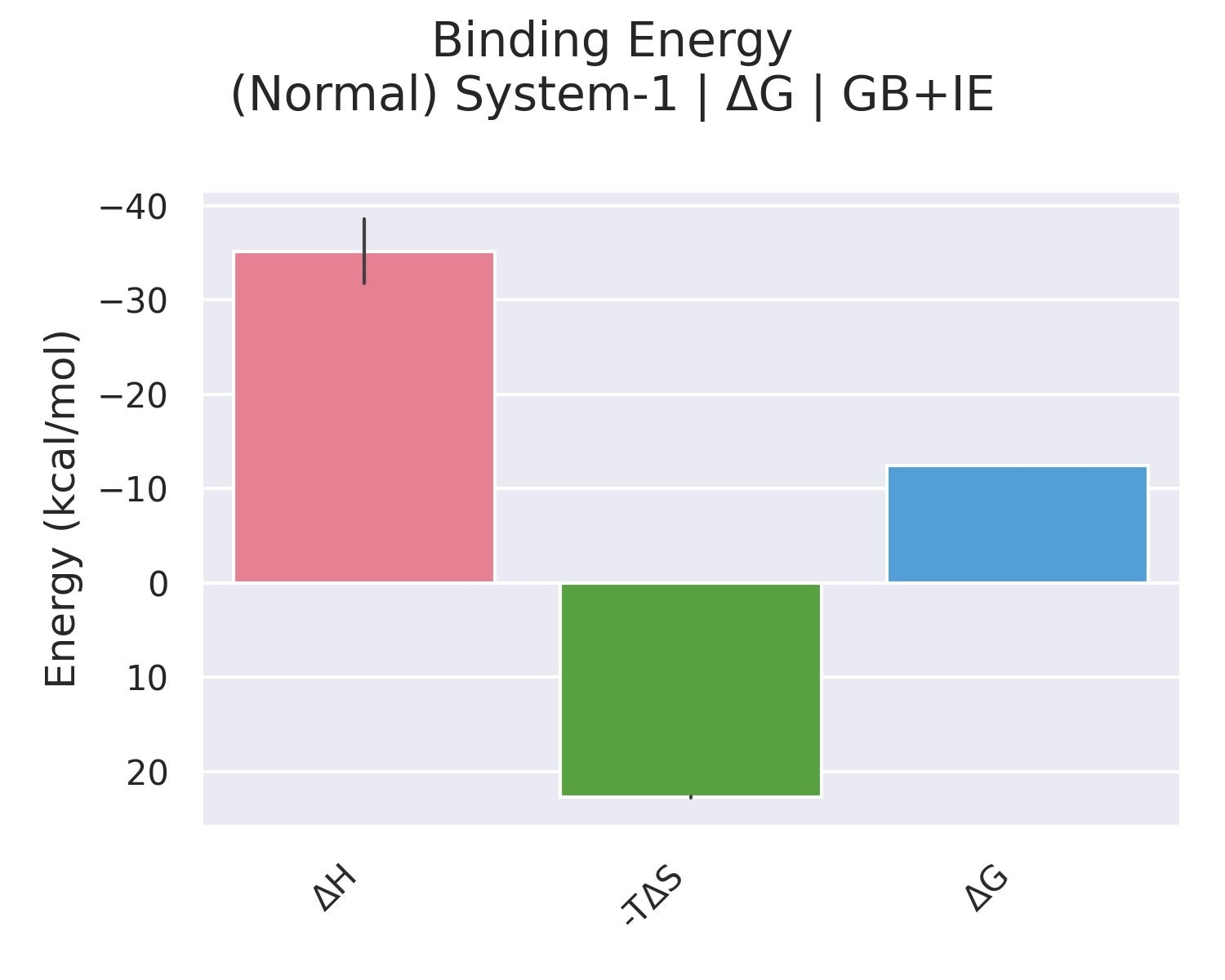
4. Binding (ΔG) 部分:最终结合自由能
这一层级将焓与熵直接相加:
- GB+IE:
\Delta G = \Delta H_{\text{GB}} + (-T\Delta S)_{\text{IE}} - PB+IE:
\Delta G = \Delta H_{\text{PB}} + (-T\Delta S)_{\text{IE}}
输出的正值或负值直接指示结合亲和力:数值越负,结合越强。一般研究中会同时报告 GB 和 PB 的结果,并优先考虑 PB 作为更可靠的估值。
5. Decomposition 部分:残基级能量贡献
当输入文件中包含 &decomp 段落且 idecomp=2 时,gmx_MMPBSA 会将 Delta 能量进一步分解到单个残基上。idecomp=2 的含义是:基于残基进行分解,并将 1-4 相互作用按类型归类(1-4 EEL 加到 EEL,1-4 VDW 加到 VDW),以保证每个残基的总贡献更直观。
Decomposition 结果同样区分 GB 与 PB,并在 Complex、Receptor、Ligand、Delta 四个分类下显示每个残基的两种贡献类型:
- TDC(Total Decomposition Contribution):残基的总能量贡献,包含主链和侧链原子。
- SDC(Sidechain Decomposition Contribution):仅来自侧链原子的贡献。
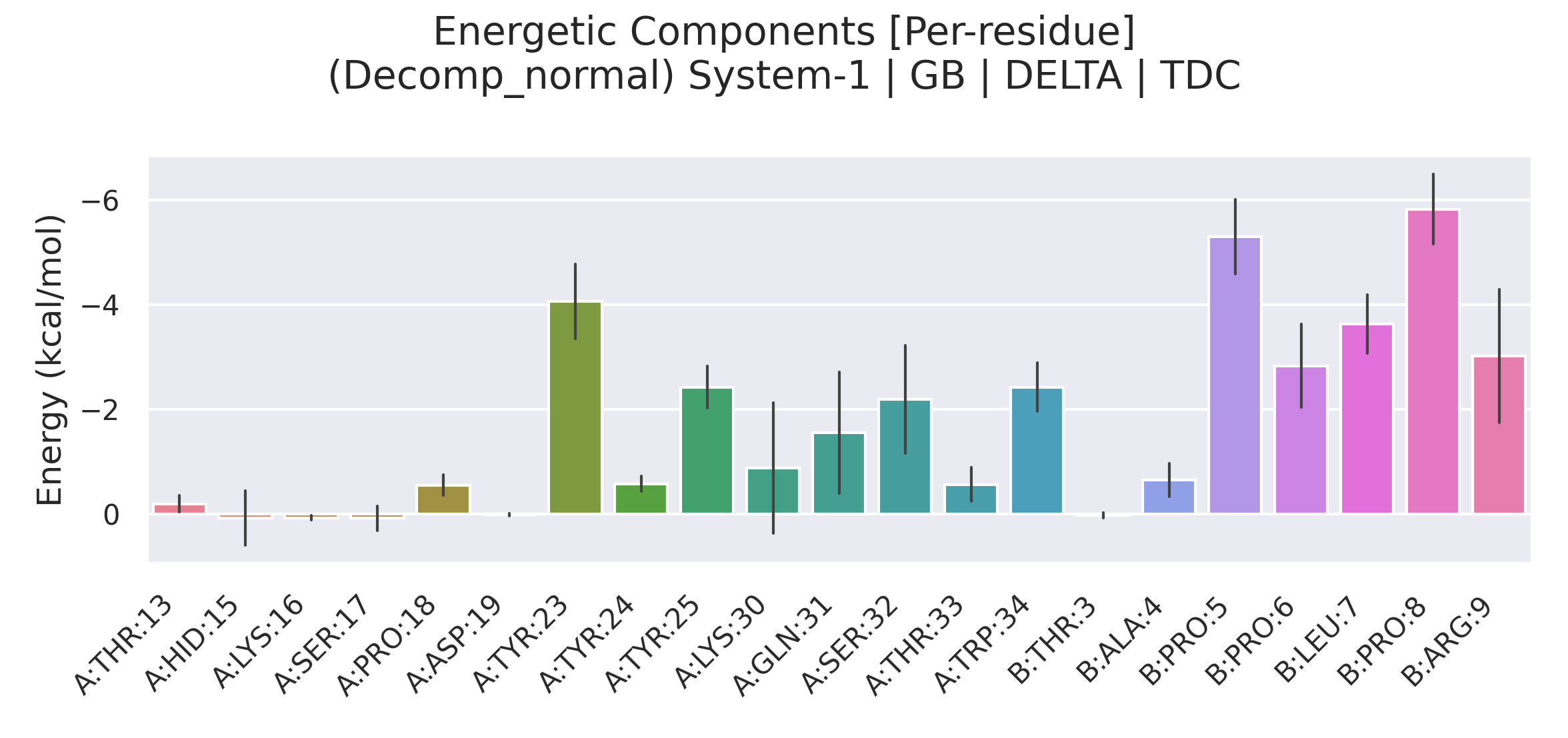
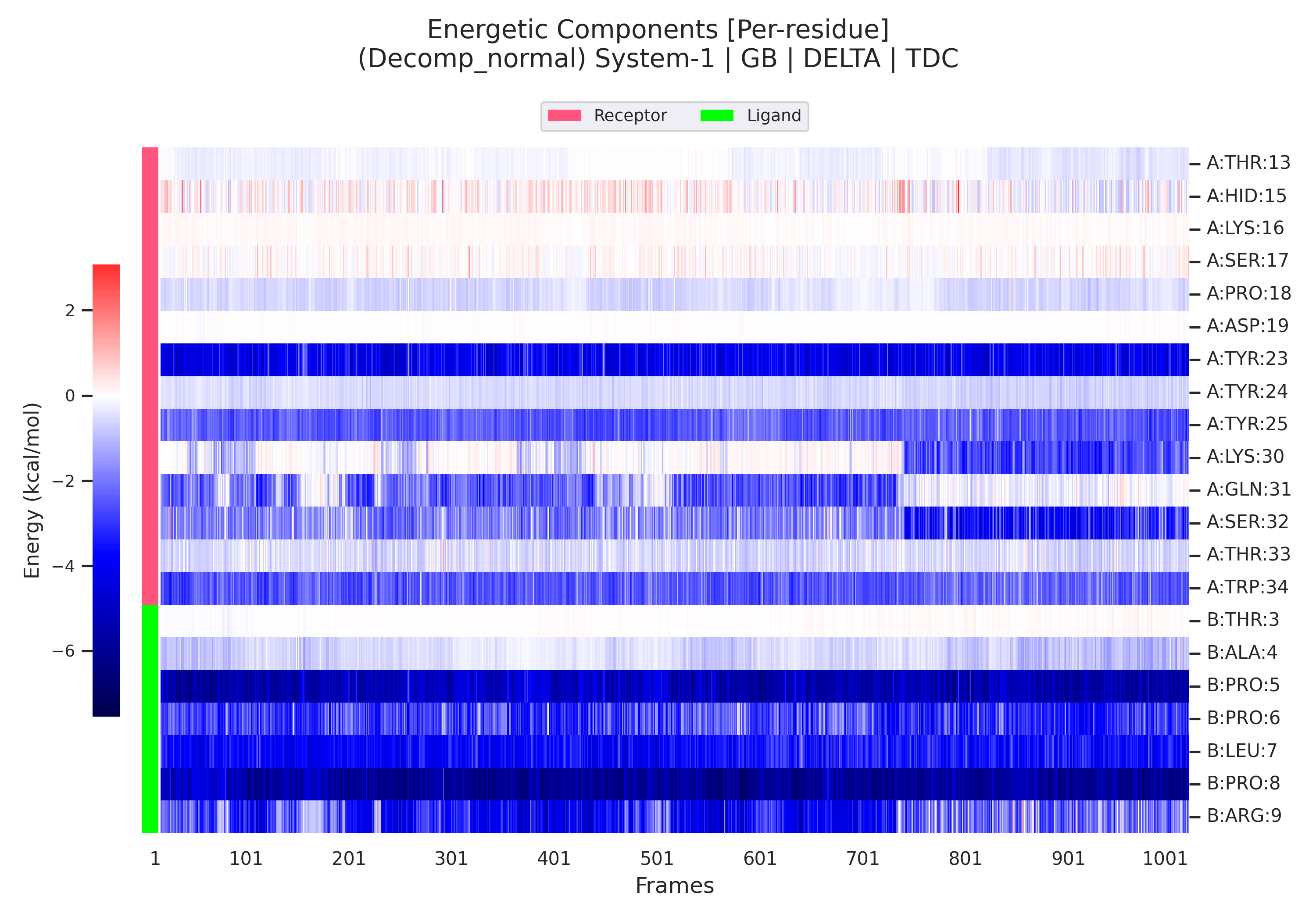
Delta 下的 TDC 和 SDC 直接展示了每个残基对结合能的净贡献(单位 kcal/mol)。通常意义下,贡献低于 -1 kcal/mol 的残基被视为热点(hotspot),而具有较大正值贡献的残基可能对结合不利。
通过对比 TDC 与 SDC,可以判断某一残基的结合贡献主要来自侧链还是主链。例如,若某残基的 SDC 接近 TDC,则贡献几乎全由侧链提供;若 SDC 较小而 TDC 较大,则主链氢键或静电作用可能占主导。
6. 结果解读示例与注意事项
分析时建议读者按照以下逻辑进行:
- 首先查看 Binding (ΔG) 下的数值,确认计算是否给出合理的负值(通常 -5 至 -15 kcal/mol 为常见的小分子结合自由能)。
- 进入 Enthalpy → Delta,观察 ΔVDWAALS 和 ΔESURF 是否为负,ΔEEL 是否为负而 ΔEGB 为正。净静电贡献(ΔEEL + ΔEGB)在介电常数设置下可能为微弱有利或轻微不利,具体取决于体系极性。
- 查看 Entropy 的 -TΔS 值,如果绝对值很大且为正,则说明熵损失显著,结合可能由焓驱动。
- 利用 Decomposition → Delta TDC 找出贡献显著的残基,并与实验突变数据或相互作用指纹进行对照。
- 结合 GB 和 PB 的趋势一致性评估模型稳健性。若两者符号甚至量级差异过大,应检查参数(如 PB 网格大小、表面张力系数等)是否合理。
最后,所有能量数值均是平均值,并伴有一个标准偏差(可通过 gmx_MMPBSA_ana 或直接查看 FINAL_RESULTS_MMPBSA.dat 获取其中的详细数据)。当标准偏差远小于平均值时,结果才具有统计意义。
最后,Enthalpy 提供气相与溶剂化对结合的驱动信息,Entropy 补充构型限制的损耗,Binding 给出最终的结合强度,Decomposition 则直接定位关键残基。这是各个部件最主要的功能

