在使用 gmx_MMPBSA 完成结合自由能计算后,gmx_MMPBSA_ana 会输出一个树形结构的结果汇总。该结构严格按照热力学函数的构成进行组织,涵盖了焓变(ΔH)、熵变贡献(-TΔS)、结合自由能(ΔG)以及基于残基的能量分...
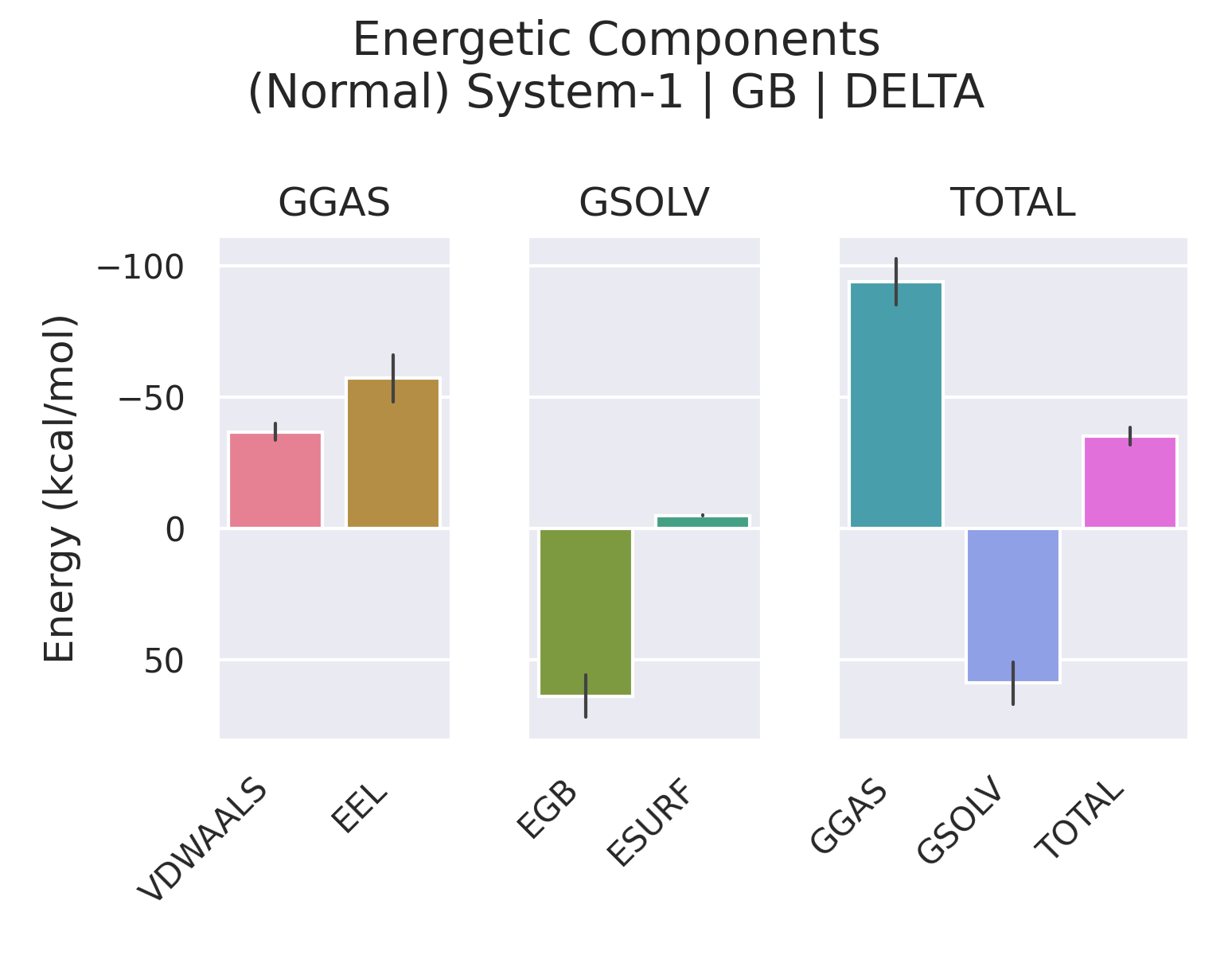


在使用 gmx_MMPBSA 完成结合自由能计算后,gmx_MMPBSA_ana 会输出一个树形结构的结果汇总。该结构严格按照热力学函数的构成进行组织,涵盖了焓变(ΔH)、熵变贡献(-TΔS)、结合自由能(ΔG)以及基于残基的能量分...
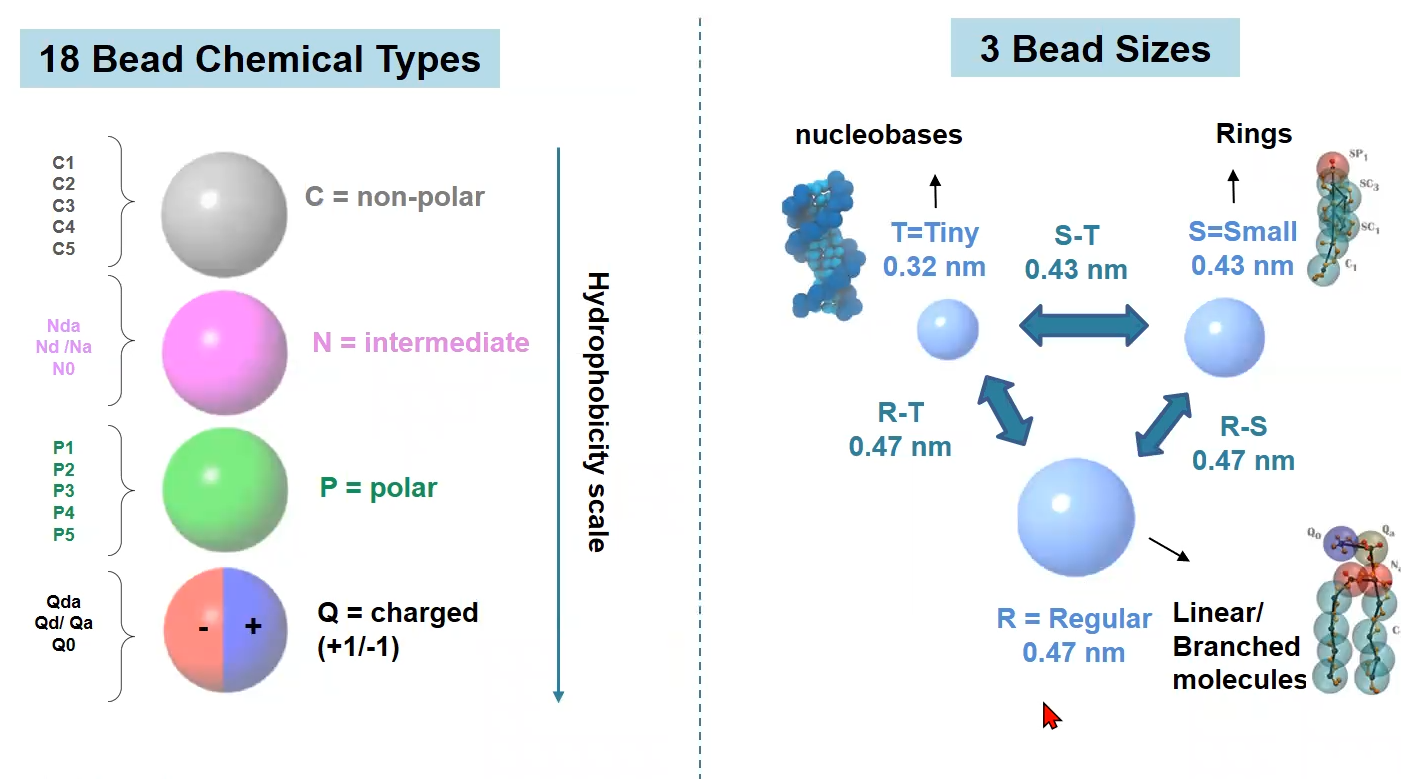
前言 在计算生物物理与材料科学的领域,分子模拟的空间与时间尺度始终是一对核心矛盾。全原子分子动力学(AA-MD)能够提供原子级别的更高的精度,但同时计算成本使得模拟超过微秒量级、或超过数十万原子的体系变...
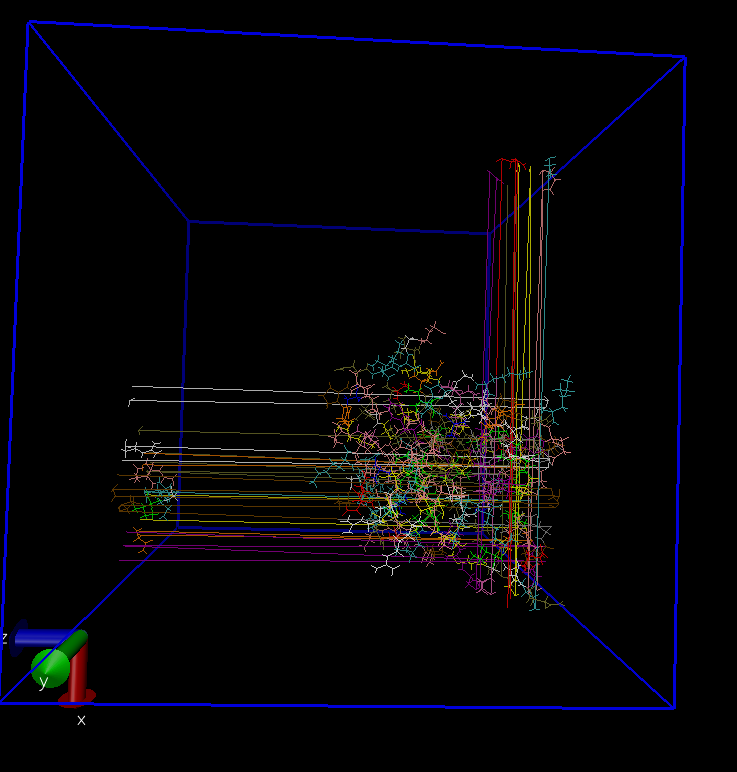
前言 在gromacs动力学中,原始轨迹文件(.xtc/.trr)因周期性边界条件(PBC)和整体平动/转动的存在,无法直接用于定量分析或可视化。本文介绍三类核心预处理方法,分别针对连续轨迹、结构叠合和可视化需求,并明...
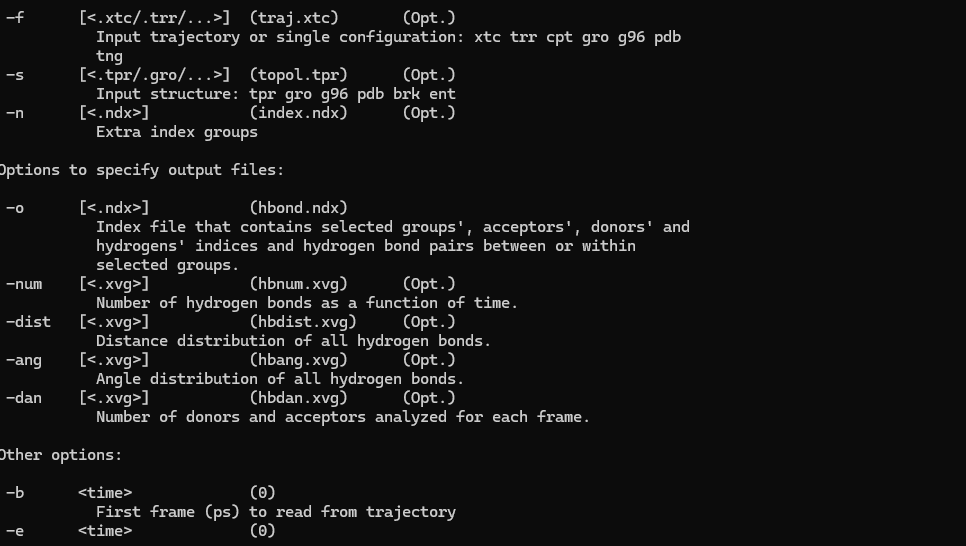
引言:一次意外的发现 “为什么我的GROMACS没有gmx hbond中的-life参数?” 当我在Windows终端中输入gmx hbond -h,仔细翻看帮助文档中每一个参数,却始终找不到期待已久的-life选项时,一种困惑油然而生。氢键寿命...